Спинальная атрофия мышц относится к тяжелым патологиям, подразумевающим нарушения двигательных функций. Различают четыре разновидности заболевания, среди которых самой неблагоприятной считается спинальная амиотрофия Верднига-Гоффмана, развивающаяся в младенческом и детском возрасте. Этот вид патологии имеет наследственный характер и не поддается излечению, а применяемые методики могут лишь немного облегчить состояние больного. По каким же признакам распознается заболевание, и что делать в случае его выявления?
Характеристика патологии
Спинальные мышечные атрофии, или СМА, подразумевают поражение нейронов спинного мозга, отвечающих за двигательные функции мускулов. Больше всего обычно страдает мускулатура ног и шеи, а вот мышцы верхних конечностей поражаются реже. У больных наблюдаются проблемы с передвижением, глотанием, удержанием головы, но при этом сохраняется чувствительность, и нет задержек в умственном развитии. Но если при остальных формах СМА у больных есть шанс дожить до старости, хоть и с инвалидностью, то при амиотрофии Верднига-Гоффмана максимальная продолжительность жизни не превышает 30 лет.

Патология встречается довольно редко – в одном случае из 80-100 тысяч. Но вот носителей гена, отвечающего за развитие аномалии, гораздо больше. Болезнь наследуется по рецессивно-аутосомному типу, и чтобы у ребенка проявилась амиотрофия Верднига-Гоффмана, оба родителя должны быть носителями гена. Хотя и в этом случае вероятность заболевания у малыша составляет только 25%. Причиной болезни является исключительно генетическая предрасположенность, никакой связи СМА с травмами, инфекциями и другими факторами не обнаружено.

Проявления болезни
Специалисты выделяют три формы СМА Верднига-Гоффмана, отличающиеся между собой сроками проявления и типичными признаками.
Таблица. Формы СМА Верднига-Гоффмана
| Форма заболевания | Сроки проявления | Продолжительность жизни |
|---|---|---|
| Младенческая (СМА I типа) | С рождения до 6 мес. | От 6 мес. до 3 лет. |
| Ранняя детская (СМА II типа) | 7-10 мес. | 14-15 лет. |
| Поздняя (СМА III типа) | От 1,5 до 2,5 лет. | 20-30 лет. |
Все указанные формы патологии объединяет отсутствие каких-либо умственных и чувствительных нарушений, зато клиническая картина имеет значительные различия.

Младенческая форма
При патологии I типа первые симптомы заметны уже при рождении ребенка: он появляется на свет с вялыми парезами, издает очень слабые крики, глубокие рефлексы отсутствуют. Для специалиста не составляет труда определить мышечную гипотонию, что позволяет с первых дней установить наличие атрофии Верднига-Гоффмана. Такие дети вяло сосут грудь, плохо глотают молоко, часто захлебываются. Даже движения языка затруднены, и при внимательном рассмотрении можно увидеть на нем непроизвольные сокращения мышц – мелкие волнообразные движения. Все это вызывает сложности с кормлением, ведь пища может проникнуть в дыхательные пути и вызвать гибель ребенка.

Кроме указанных симптомов наблюдается парез диафрагмы и деформации скелета: у ребенка может быть искривлен позвоночник, вдавлена или, наоборот, остро выступающая грудная клетка, вывернуты суставы. Дети с таким диагнозом сильно отстают в моторном развитии по сравнению со сверстниками. Они не могут удерживать голову, переворачиваться со стороны в сторону, тянуться за привлекающим внимание предметом и принимать сидячее положение. При этом мимику лица и функции глазных мышц болезнь не затрагивает, и эмоции, которые выражает ребенок, не искажаются.
К сведению: у ограниченного числа детей с амиотрофией Верднига-Гоффмана двигательные навыки все же проявляются, хоть и с большим опозданием, но затем через короткое время регрессируют.
СМА первого типа часто сопровождается другими врожденными патологиями:
 Читайте также:
Читайте также:
- дисплазией суставов таза;
- гидроцефалией головного мозга;
- гемангиомами;
- пороком сердца.
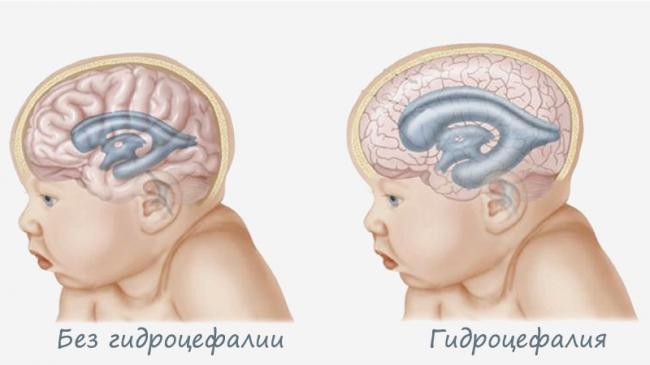
Развивается заболевание очень быстро, и летальный исход в большинстве случаев наступает в течение полугода после рождения. Некоторые малыши доживают до 2-3 лет. Причиной смерти, как правило, служит тяжелая форма сердечной и дыхательной недостаточности.
Ранняя форма
При втором типе амиотрофии дети в первые шесть месяцев своей жизни развиваются абсолютно нормально, без каких-либо настораживающих признаков. Некоторые в полгода даже начинают вполне активно подниматься на ноги и передвигаться вдоль кроватки или манежа. Первым симптомом является мышечная слабость, которая может развиваться постепенно или резко возникать на фоне любого детского заболевания, различных инфекций. Происходит это, как правило, на 7-10 месяце жизни малыша.

Функции мускулов сначала нарушаются в нижних конечностях, вследствие чего ребенок все хуже ползает, с трудом встает на ножки. Далее поражение распространяется все выше, что приводит к снижению глубоких рефлексов, появлению тремора пальцев, произвольных мускульных сокращений в языке, нарушению работы органов дыхания. Протекает данная форма патологии менее интенсивно, чем младенческая, и большинство больных доживает до подросткового возраста, хотя качество жизни все это время остается очень низким. Такие дети не могут себя обслуживать самостоятельно и без посторонней помощи им не обойтись. Причиной смерти обычно служат нарушения в работе дыхательных органов, а также различные инфекции, которые нередко поражают ослабленный болезнью организм.
Поздняя форма
Данную форму СМА специалисты считают наименее тяжелой, хотя и с ней человек может прожить максимум до 30 лет. Типичные проявления возникают чаще всего, когда ребенку исполнится 1,5-2 года. До этого времени физическое развитие происходит без малейших нарушений, малыш отлично ходит, бегает, проявляет нормальную для своего возраста активность. Лишь у ограниченного числа больных могут наблюдаться задержки с развитием моторики или излишняя медлительность.

Первые симптомы слабо выражены:
- ребенок быстрее устает;
- снижается координация движений, малыш чаще падает при беге и ходьбе;
- часто наблюдается вялость.
С прогрессированием болезни появляются другие признаки, более характерные:
- меняется походка, ребенок выше поднимает колени во время ходьбы;
- усиливается мышечная слабость;
- наблюдается мелкое дрожание пальцев рук;
- возникают произвольные спазмы языка, затруднения с глотательными функциями;
- развиваются деформации костей и суставов, особенно выражено это в грудной клетке.
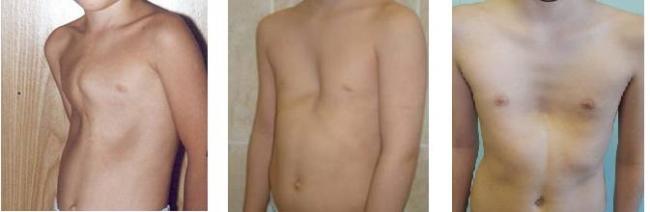
Все эти процессы отличаются довольно медленным развитием, и способность ходить сохраняется примерно до 8-10 лет. В дальнейшем передвижение возможно лишь в инвалидной коляске, но способность к самообслуживанию полностью не утрачивается еще несколько лет. При наличии поддерживающей терапии люди с таким диагнозом доживают до 25-30 лет.
Диагностика заболевания
Синдром врожденной гипотонии мышц характерен не только СМА, но и ряду других патологий, например, ДЦП, амиотрофическому склерозу, миопатиям разных форм. Поэтому визуальный осмотр, а также информация о сроках появления симптомов и динамики их развития не помогут дифференцировать амиотрофию Верднига-Гоффмана. Исключить наличие других спинальных патологий позволяет проведение КТ или МРТ, но для определения СМА этих исследований недостаточно.
Основным диагностическим методом при подозрении на спинальную атрофию мышц является электронейромиография, или ЭНМГ. Данный способ предназначен для анализа работы нервно-мышечной системы, в частности, прохождения нервных импульсов и реакции на них. Для подтверждения диагноза также назначается биопсия мышечных тканей и ДНК-исследования.
 Читайте также:
Читайте также:
Важно! Людям, у которых в семьях уже наблюдались случаи заболевания СМА, рекомендуется пройти генетическое исследование на наличие SMN-гена, ответственного за нарушения развития мышц. Беременным женщинам проводят дородовый ДНК-анализ плода, и если диагноз подтвердится, это является серьезным показанием к прерыванию беременности.
Поддерживающая терапия
СМА Верднига-Гоффмана неизлечима, поэтому больным оказывается поддерживающая терапия, которая направлена на облегчение симптомов заболевания. Основной акцент делается на улучшение метаболизма в пораженных мышечных и нервных волокнах, что позволяет замедлить прогрессирование патологии. Для этого применяют медикаментозные средства нескольких групп: нейрометаболиты, препараты для облегчения нервно-мышечной передачи, препараты для улучшения трофики тканей и кровообращения.
Чаще всего назначаются:
- «Церебролизин»;
- «Ноотропил»;
 Раствор для инъекций «Ноотропил»
Раствор для инъекций «Ноотропил» - «Аминалон»;
- никотиновая кислота;
- «Скополамин»;
- «Нейромидин».
 Таблетки «Нейромидин»
Таблетки «Нейромидин»
Вид препаратов, дозировки, длительность приема определяются лечащим врачом по результатам обследования и с учетом возраста ребенка. Помимо лекарств показана лечебная физкультура, физиотерапевтические процедуры, мягкий массаж. При усиленной деформации позвоночника применяются ортопедические корсеты и бандажи.
Видео — Спинальная амиотрофия Верднига-Гоффмана
Мышечная атрофия — не единственная патология двигательной системы, что передается генетически. О том, какие еще существуют наследственные заболевания позвоночника, и как с ними бороться, можно прочитать на нашем сайте.